How to predict many protein structures with AlphaFold2 at-scale in Azure Machine Learning
Back in December, I published a couple posts here (post 1 and post 2) about using AlphaFold2 and RoseTTAFold to predict the receptor binding domain (RBD) of the SARS-CoV-2 Omicron variant. The motivation for this was due to fact that the actual structure of the RBD wasn’t yet available. (It is now. PDB: 7T9J) AlphaFold2 did an excellent job at predicting this structure, which has me wanting to predict even more structures.

But how would we go about generating tons of structures at once? Let’s say you want to generate predicted structures for a whole list of sequences. We could sequentially run each sequence through AlphaFold2 and collect the result, but this would be time-consuming and manual.
Normally, Azure Machine Learning is used for…well…machine learning. So, its toolkit is often used to scale-up machine learning training tasks. However, we can use these tools for virtually any computational task.
In this post, I will cover how to utilize he power of Azure Machine Learning’s HyperDrive system to automate and distribute the AlphaFold2 folding tasks to generate structures in parallel.
Note: We’ll be using ColabFold, a more accessible wrapper for AlphaFold2 and RoseTTAFold for running predictions in a notebook-style environment.
Input Data
We can provide our input sequences as a single FASTA file. For this demo, I have a sequences.fasta file with 4 SARS-CoV-2 receptor binding domain amino acid sequences.

Getting Started
Assuming you already have an Azure Machine Learning Workspace with a Compute Instance up and running, open up JupyterLab and create a new Python notebook. (If you don’t have a Workspace already, follow this tutorial.)
Pro tip: You can just clone my full notebook from the GitHub repo listed in Resources below and open it in JupyterLab.
Once in the notebook, make sure to load in your Workspace configuration:
Creating an Inference Cluster
Inference clusters are usually used for distributed machine learning model training. This time, though, we’re going to use one for distributed protein folding tasks.
Create your cluster using the Python SDK with the following script:
Note: We’re using GPU-enable virtual machines in the cluster (NC6’s, line 13).
Run Configuration
Custom Environments
Compute environments are important as they help define the required library dependencies for model training. In this case, I’ve created a Docker image that contains ColabFold (with AlphaFold2) and all the required dependencies to make it run. We’ll define the environment by telling it to pull this image onto each of the nodes of the compute cluster (lines 6–9 below).
We also tell the system what the input arguments are (in this case, prediction settings), what script to run to do the work, etc.
Hyperdrive
HyperDrive is the built-in distribution system in Azure Machine Learning. Normally, this system would aid in hyperparameter tuning by picking parameter sets and spinning off individual machine learning training processes to perform parallel model training using these parameters.
For our protein structure prediction, we’ll just use HyperDrive to manage the selection of different sequence IDs that will be sent to the individual prediction nodes in the cluster (line 22 in the script below).
Setting up the Prediction Script
Normally, in a distributed training experiment, we would create a train.py file that would get executed on each compute node to train a model. Here, we’ll create a similar predict.py file that will take in the arguments from the HyperDrive engine (the sequence ID to be folded and some input settings) and execute on the compute node from which it’s running.
This script will tell AlphaFold2 to output the files in the ./outputs/ directory, which means we’ll be able to grab the results once all the predictions are done. Also, writing data to this directory means that we can also see the files from the Workspace UI.
Submit the Experiment
Finally, we can submit the HyperDrive experiment, which will distribute one sequence to each of the available compute nodes, predict the folded structure, and then complete.
This will display a live panel in your notebook that will show the runs that have been queued/are running/have finished. You can also see the same information from the Workspace UI.
This will create a Parent Run, under which will be 4 Child Runs (one for each of our input sequences).

Once a Child Run is complete (in under half an hour), you can navigate into the Outputs + logs and see the stdout of the run and also browse the results of the prediction run. You’ll find the output PDB file of the predicted structure and some metrics under ./outputs/predicted_structures/.

Collecting all the Results
Once all of your predicted structures have been created, we can grab the output PDBs (and other results) from each of the experiment runs back in JupyterHub so that we can interactively compare our structures.
This will create a ./results folder with subfolders for each of the sequences we ran in this experiment.
Rendering PDBs in the Notebook
Thanks to the py3dmol package, we can now render 3D structures right from the notebook. First, we’ll crawl the ./results folder for PDB files and then pass those in as options to the interactive viewer.
This will show a 3D viewer window where you can toggle between the 4 predicted structures and multiple viewing styles (cartoon, surface, stick, etc.).
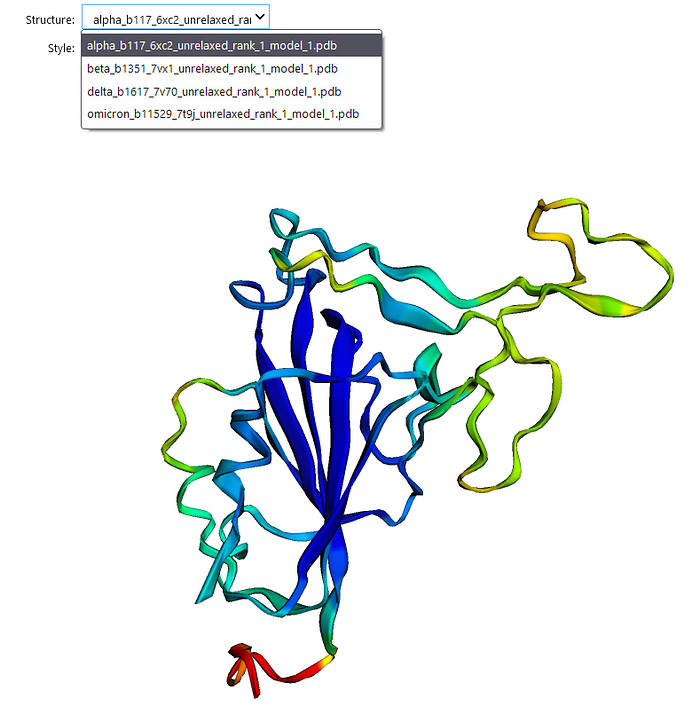
While not as full-featured as PyMol or other desktop applications, this is a nice way to get a quick peek at your predicted structures right in the notebook.
Conclusion
Deep learning-based protein structure prediction tools like AlphaFold2 and RoseTTAFold have huge implications for helping us understand the shapes of various proteins for which experimentally-derived structures do not yet exist. Running single sequences through these systems is time-consuming. So when we need to scale-up and predict the structures of many sequences, we need to turn to more scalable tools to do so.
Azure Machine Learning, though made for machine learning training, can be used to distribute computationally-intensive workloads like this one in just a few straightforward steps.
Individually, each of these example structures would’ve taken ~28 minutes to complete. Using HyperDrive, we can make all of the structures in parallel in approximately the same amount of time.
Resources
- All code for this post is available on GitHub at https://github.com/colbyford/azureml-alphafold2
- ColabFold GitHub repo: https://github.com/sokrypton/ColabFold
- Post: Predicted Protein Interactions of the SARS-CoV-2 B.1.1.529 Variant with Neutralizing Antibodies
- Post: Protein Structure Prediction of the new B.1.1.529 SARS-CoV-2 Spike Variant with AlphaFold2
- Preprint: Predictions of the SARS-CoV-2 Omicron Variant (B.1.1.529) Spike Protein Receptor-Binding Domain Structure and Neutralizing Antibody Interactions
