Protein Structure Prediction of the new B.1.1.529 SARS-CoV-2 Spike Variant with AlphaFold2
It’s been a bit since we’ve heard much about a new COVID variant. With people starting to get their boosters and the holidays in full-swing, this seems like an inopportune time to hear about a new, “mega-mutated” variant: B.1.1.529.
B.1.1.529 was first uploaded to GISAID on November 22nd (accession: EPI_ISL_6752027) by a team of researchers from the Botswana Harvard HIV Reference Laboratory. It contains 60 amino acid mutations from the reference genome, 37 of which are in the Spike protein.

Given that the Spike protein is what vaccines (and therefore our antibodies) are based upon, we naturally want to understand if any of these mutations might affect the receptor binding domain (RBD) of the Spike protein. The RBD is from approximately residues 335 to 525 of the S protein.
There are 15 mutations in the RBD, with zero deletions or insertions.
The 3D structure for the Spike protein of B.1.1.529, at the time of writing, has not been determined. So, I turned to AlphaFold2, a deep learning-based model, to give it guess.
I used ColabFold, which provides Jupyter notebooks for AlphaFold2 and RoseTTAFold that you can run in Google Colab or any other .ipynb-compatible notebook application with a GPU.
Many neural net-based structural prediction tools work by finding homologous sequences and then checking to see if those sequences have known structures. This provides a scaffold on which to overlay the query sequence and then the model can focus on the parts that don’t match the reference structure.
AlphaFold2 RBD Results
The AlphaFold2-generated structure is shown below in blue with mutated residues shown in red. Note: This is in comparison with a reference structure (PDB: 6VSB, chain A) in green.

It appears that AlphaFold2 has made a decent guess as to the overall 3D structure, showing that this set of mutations isn’t causing a huge conformational change in the RBD. This is either due to AlphaFold2 overusing a homolog as the prediction or maybe it’s actually right. Who knows?
Note that some of these mutated residues change to much longer side-chained or differently-charged amino acids. For example, there are two “to lysine” mutations: N440K and T478K (i.e., from polar, smaller side chain residues to a positive-charged, longer side chain residue). These types of changes may have an effect on the binding affinity between the RBD and an antibody, either by changing the surface charge on the protein or by blocking a tighter antibody interaction.
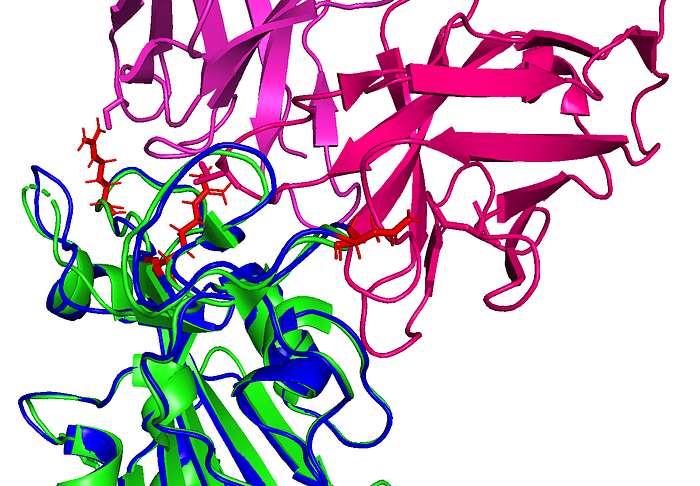
In this figure showing a RBD-antibody interaction, note how mutations Q498R, N501Y, and S477N from the predicted B.1.1.529 structure may affect binding position of the antibody, causing it to bind less effectively. In other words, these longer/larger side chains may increase the distance between the Fab paratope of the antibody and the epitope of the RBD.
How Bad Is It, Doc?
In short, it’s hard to say. Mutations in the RBD open the door to reinfection with the virus. This is due to the virus evading existing antibodies because they fail to bind sufficiently to the RBD. In terms of vaccinations, the same applies. We may need a B.1.1.529-based booster to combat this variant if it becomes widespread in the future.
In terms of health, some of the other mutations (outside of the RBD) may enable the virus to enter our cells more easily (by increasing the interaction between the human ACE2 receptor and the Spike protein). Some mutations may increase the pathogenicity of the virus, making it more harmful to us if we become infected.
Predictions Leading to Actions
Determining the actual structure of a protein takes a lot of time and effort. Also, quantifying protein-protein interactions (like Spike-to-antibody interactions) are also difficult to derive in vitro. Thus, predictive tools like AlphaFold2 and RoseTTAFold are important for quickly identifying risks like COVID-19 variants.
Using these tools, we can get an in silico understanding of how certain mutations may affect us in real life. From vaccine efficacy to virulence to pathogenicity, computational tools will provide value in the seemingly ever-evolving COVID-19 pandemic.
Resources
All code, sequences, and results are available on GitHub here: https://github.com/colbyford/SARS-CoV-2_B.1.1.529_Spike-RBD_Predictions
If you use any of this work, code, insights, or results, please cite:
Colby T. Ford. (2021). Predictions of the SARS-CoV-2 B.1.1.529 Variant Spike Protein Receptor Binding Domain Structure and Neutralizing Antibody Interactions (v1.0.0) [Data set]. Zenodo. https://doi.org/10.5281/zenodo.5733161
Stay curious.
